Continuous- versus Segmented-Flow Microfluidic Synthesis in Materials Science



Abstract
1. Introduction
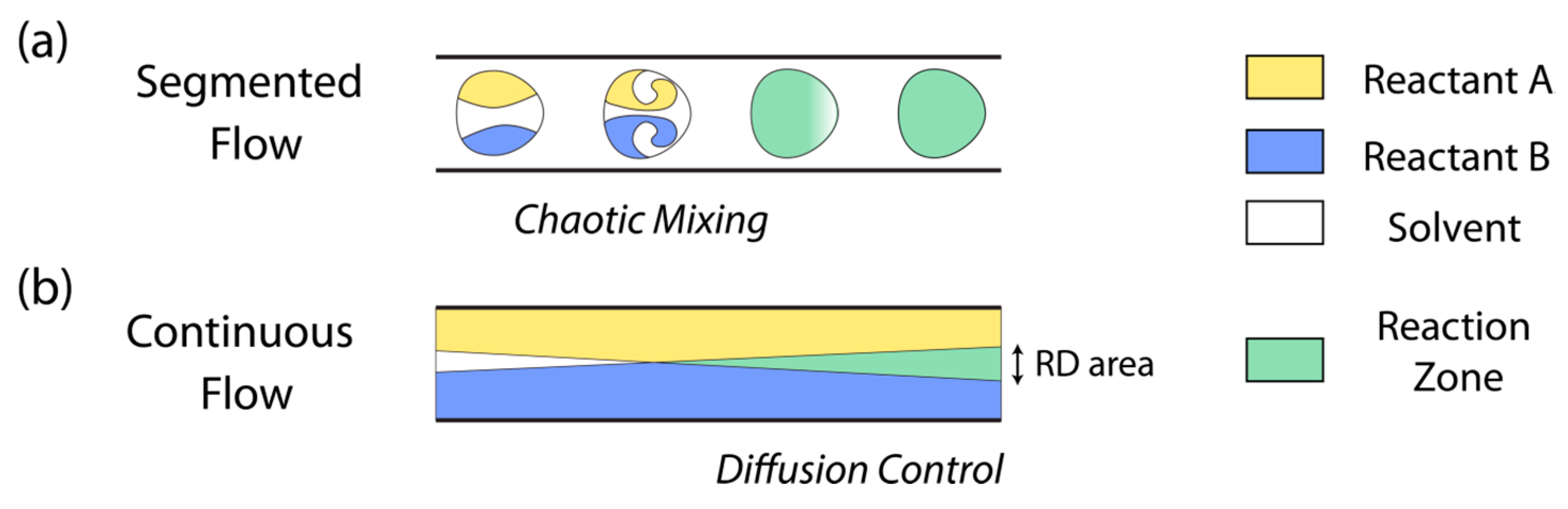
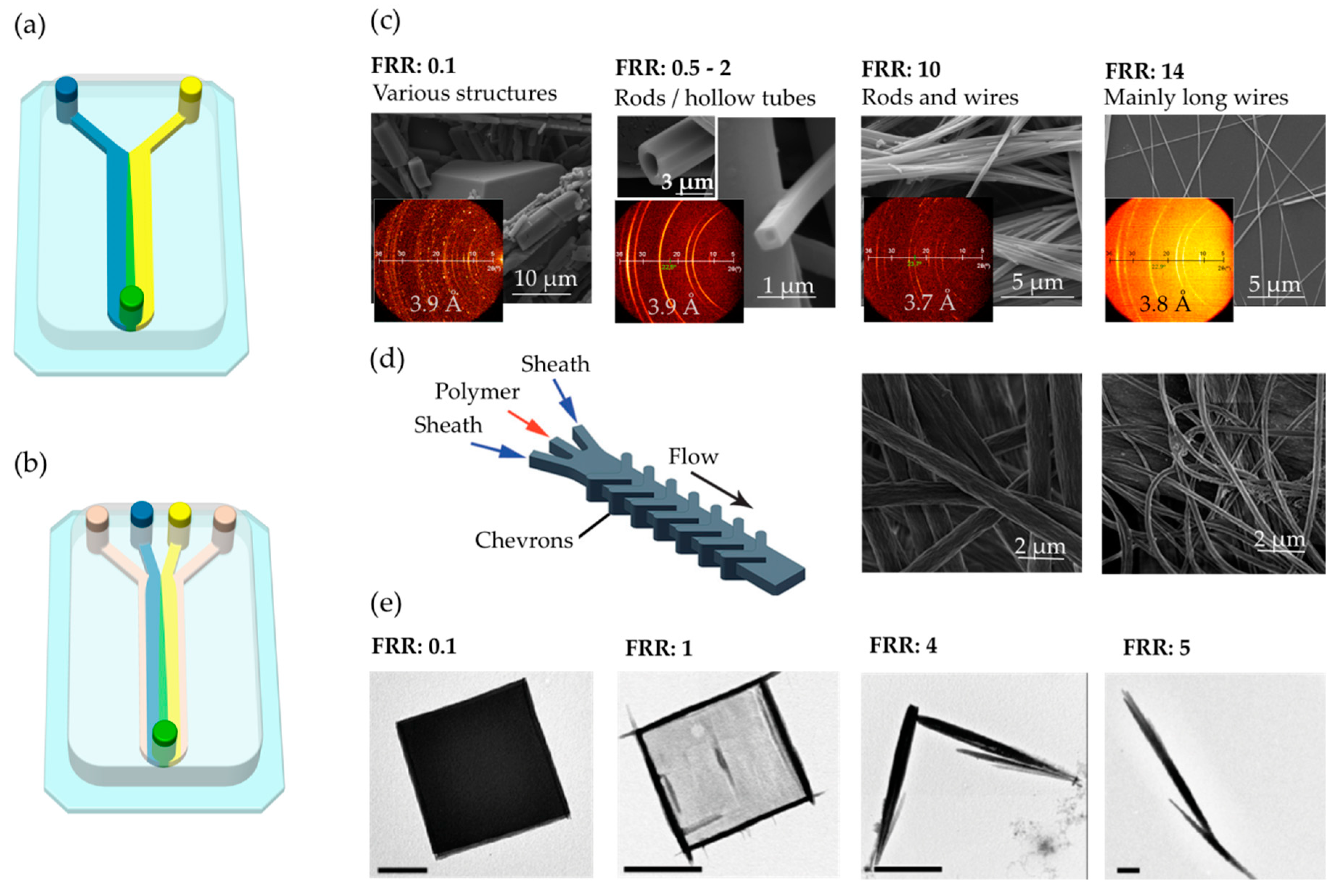
2. Continuous Flow
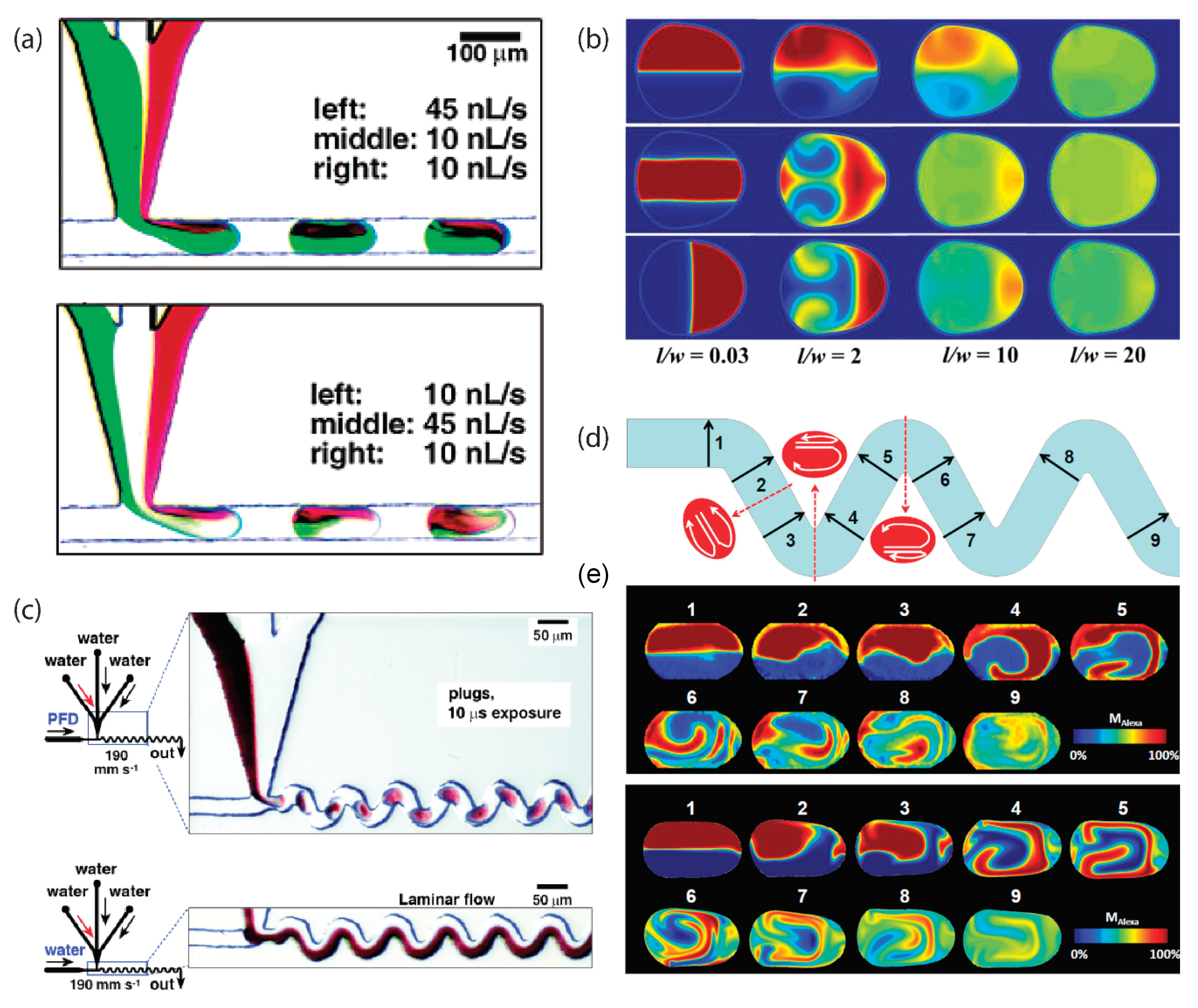
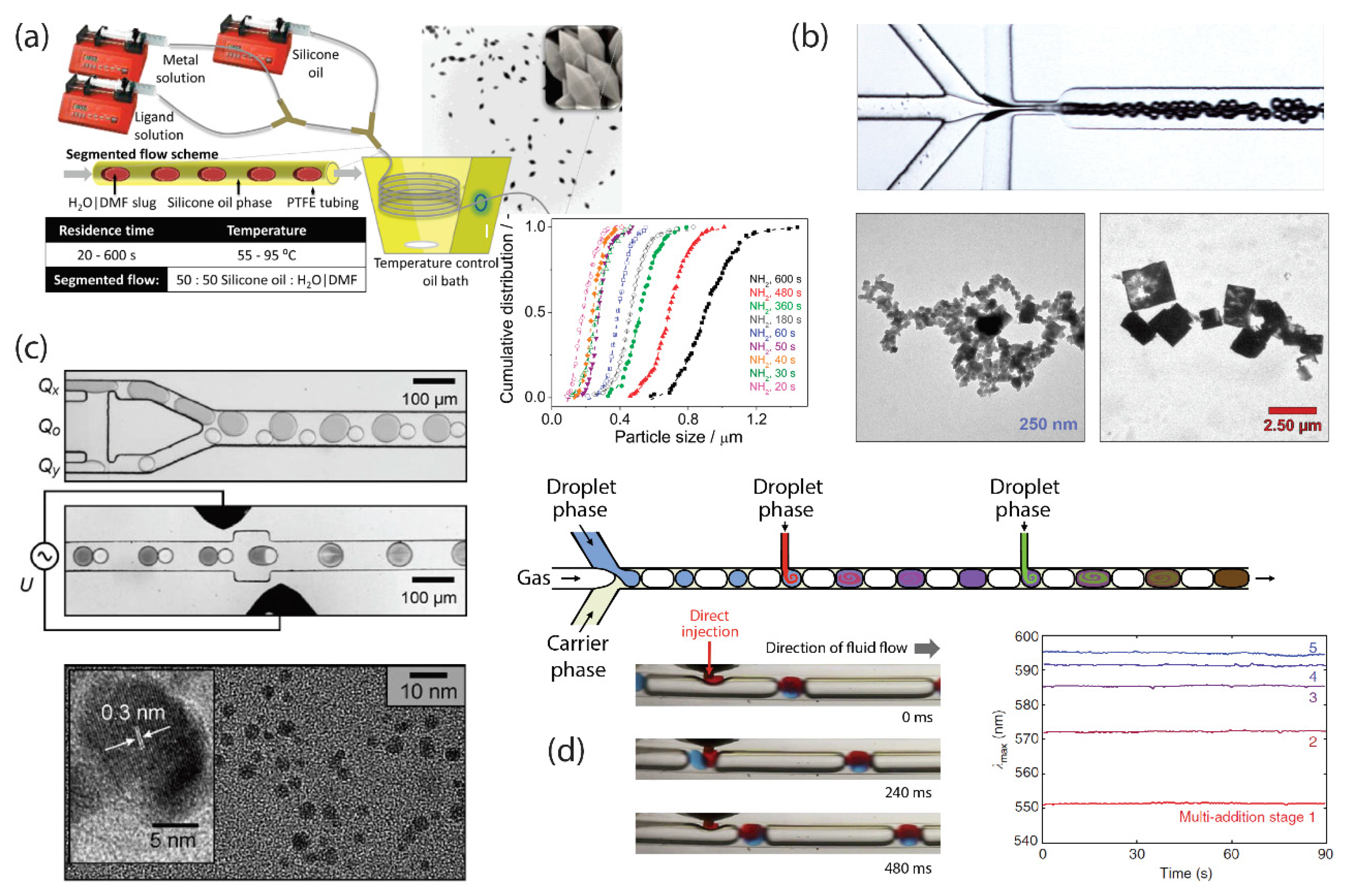
3. Segmented-Flow Microfluidics
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Ehrfeld, W.; Hessel, V.; Löwe, H. Microreactors: New Technology for Modern Chemistry; Wiley-VCH: New York, NY, USA, 2000; ISBN 9783527295906. [Google Scholar]
- Jensen, K. Chemical kinetics: Smaller, faster chemistry. Nature 1998, 393, 735–737. [Google Scholar] [CrossRef]
- Squires, T.M.; Quake, S.R. Microfluidics: Fluid physics at the nanoliter scale. Rev. Mod. Phys. 2005, 77, 977–1026. [Google Scholar] [CrossRef]
- Nguyen, N.; Wereley, S. Fundamentals and Applications of Microfluidics; Artech House: Boston, MA, USA, 2006; ISBN 1580539726. [Google Scholar]
- Purcell, E.M. Life at low Reynolds number. Am. J. Phys. 1977, 45, 3–11. [Google Scholar] [CrossRef]
- Elvira, K.S.; I. Solvas, X.C.; Wootton, R.C.R.R.; DeMello, A.J. The past, present and potential for microfluidic reactor technology in chemical synthesis. Nat. Chem. 2013, 5, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Jahn, A.; Vreeland, W.N.; Gaitan, M.; Locascio, L.E. Controlled Vesicle Self-Assembly in Microfluidic Channels with Hydrodynamic Focusing. J. Am. Chem. Soc. 2004, 126, 2674–2675. [Google Scholar] [CrossRef] [PubMed]
- Dendukuri, D.; Pregibon, D.C.; Collins, J.; Hatton, T.A.; Doyle, P.S. Continuous-flow lithography for high-throughput microparticle synthesis. Nat. Mater. 2006, 5, 365–369. [Google Scholar] [CrossRef]
- Puigmartí-Luis, J. Microfluidic platforms: A mainstream technology for the preparation of crystals. Chem. Soc. Rev. 2014, 43, 2253–2271. [Google Scholar] [CrossRef]
- Thangawng, A.L.; Howell, P.B., Jr.; Richards, J.J.; Erickson, J.S.; Ligler, F.S. A simple sheath-flow microfluidic device for micro/nanomanufacturing: Fabrication of hydrodynamically shaped polymer fibers. Lab Chip 2009, 9, 3126. [Google Scholar] [CrossRef]
- Rodríguez-San-Miguel, D.; Abrishamkar, A.; Navarro, J.A.R.; Rodriguez-Trujillo, R.; Amabilino, D.B.; Mas-Ballesté, R.; Zamora, F.; Puigmartí-Luis, J. Crystalline fibres of a covalent organic framework through bottom-up microfluidic synthesis. Chem. Commun. 2016, 52, 9212–9215. [Google Scholar] [CrossRef]
- Brown, A.J.; Brunelli, N.A.; Eum, K.; Rashidi, F.; Johnson, J.R.; Koros, W.J.; Jones, C.W.; Nair, S. Interfacial microfluidic processing of metal-organic framework hollow fiber membranes. Science 2014, 345, 72 LP-75. [Google Scholar] [CrossRef]
- Cacho-Bailo, F.; Catalán-Aguirre, S.; Etxeberría-Benavides, M.; Karvan, O.; Sebastian, V.; Téllez, C.; Coronas, J. Metal-organic framework membranes on the inner-side of a polymeric hollow fiber by microfluidic synthesis. J. Memb. Sci. 2015, 476, 277–285. [Google Scholar] [CrossRef]
- Song, H.; Ismagilov, R.F. Millisecond Kinetics on a Microfluidic Chip Using Nanoliters of Reagents. J. Am. Chem. Soc. 2003, 125, 14613–14619. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.M.; Alivisatos, A.P.; Mathies, R.A. High-Temperature Microfluidic Synthesis of CdSe Nanocrystals in Nanoliter Droplets. J. Am. Chem. Soc. 2005, 127, 13854–13861. [Google Scholar] [CrossRef] [PubMed]
- Abou-Hassan, A.; Sandre, O.; Cabuil, V. Microfluidics in Inorganic Chemistry. Angew. Chem. Int. Ed. 2010, 49, 6268–6286. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Roach, L.S.; Ismagilov, R.F. Screening of Protein Crystallization Conditions on a Microfluidic Chip Using Nanoliter-Size Droplets. J. Am. Chem. Soc. 2003, 125, 11170–11171. [Google Scholar] [CrossRef]
- Lau, B.T.C.; Baitz, C.A.; Dong, X.P.; Hansen, C.L. A Complete Microfluidic Screening Platform for Rational Protein Crystallization. J. Am. Chem. Soc. 2007, 129, 454–455. [Google Scholar] [CrossRef] [PubMed]
- Sevim, S.; Sorrenti, A.; Franco, C.; Furukawa, S.; Pané, S.; de Mello, A.J.; Puigmartí-Luis, J. Self-assembled materials and supramolecular chemistry within microfluidic environments: From common thermodynamic states to non-equilibrium structures. Chem. Soc. Rev. 2018, 47, 3788–3803. [Google Scholar] [CrossRef]
- Kenis, P.J.A.; Ismagilov, R.F.; Whitesides, G.M. Microfabrication inside capillaries using multiphase laminar flow patterning. Science 1999, 285, 83–85. [Google Scholar] [CrossRef]
- Hou, X.; Zhang, Y.S.; Santiago, G.T.; Alvarez, M.M.; Ribas, J.; Jonas, S.J.; Weiss, P.S.; Andrews, A.M.; Aizenberg, J.; Khademhosseini, A. Interplay between materials and microfluidics. Nat. Rev. Mater. 2017, 2, 17016. [Google Scholar] [CrossRef]
- Fleischli, F.D.; Dietiker, M.; Borgia, C.; Spolenak, R. The influence of internal length scales on mechanical properties in natural nanocomposites: A comparative study on inner layers of seashells. Acta Biomater. 2008, 4, 1694–1706. [Google Scholar] [CrossRef]
- Puigmartí-Luis, J.; Schaffhauser, D.; Burg, B.R.; Dittrich, P.S. A Microfluidic Approach for the Formation of Conductive Nanowires and Hollow Hybrid Structures. Adv. Mater. 2010, 22, 2255–2259. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Martinez, M.; Imaz, I.; Domingo, N.; Abrishamkar, A.; Mayor, T.S.; Rossi, R.M.; Carbonell, C.; DeMello, A.J.; Amabilino, D.B.; Maspoch, D.; et al. Freezing the Nonclassical Crystal Growth of a Coordination Polymer Using Controlled Dynamic Gradients. Adv. Mater. 2016, 28, 8150–8155. [Google Scholar] [CrossRef] [PubMed]
- Puigmartí-Luis, J.; Rubio-Martínez, M.; Hartfelder, U.; Imaz, I.; Maspoch, D.; Dittrich, P.S. Coordination Polymer Nanofibers Generated by Microfluidic Synthesis. J. Am. Chem. Soc. 2011, 133, 4216–4219. [Google Scholar] [CrossRef] [PubMed]
- Jun, Y.; Casula, M.F.; Sim, J.-H.; Kim, S.Y.; Cheon, J.; Alivisatos, A.P. Surfactant-Assisted Elimination of a High Energy Facet as a Means of Controlling the Shapes of TiO2 Nanocrystals. J. Am. Chem. Soc. 2003, 125, 15981–15985. [Google Scholar] [CrossRef] [PubMed]
- Bastani, D.; Esmaeili, N.; Asadollahi, M. Polymeric mixed matrix membranes containing zeolites as a filler for gas separation applications: A review. J. Ind. Eng. Chem. 2013, 19, 375–393. [Google Scholar] [CrossRef]
- Li, J.-R.; Kuppler, R.J.; Zhou, H.-C. Selective gas adsorption and separation in metal–organic frameworks. Chem. Soc. Rev. 2009, 38, 1477. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, T.; Roberts, R.W.; Arnold, F.H.; Quake, S.R. Dynamic Pattern Formation in a Vesicle-Generating Microfluidic Device. Phys. Rev. Lett. 2001, 86, 4163–4166. [Google Scholar] [CrossRef] [PubMed]
- Marre, S.; Jensen, K.F. Synthesis of micro and nanostructures in microfluidic systems. Chem. Soc. Rev. 2010, 39, 1183. [Google Scholar] [CrossRef]
- Shestopalov, I.; Tice, J.D.; Ismagilov, R.F. Multi-step synthesis of nanoparticles performed on millisecond time scale in a microfluidic droplet-based system. Lab Chip 2004, 4, 316. [Google Scholar] [CrossRef]
- Frenz, L.; El Harrak, A.; Pauly, M.; Bégin-Colin, S.; Griffiths, A.D.; Baret, J.C. Droplet-based microreactors for the synthesis of magnetic iron oxide nanoparticles. Angew. Chem. Int. Ed. 2008, 47, 6817–6820. [Google Scholar] [CrossRef]
- Hung, L.-H.; Choi, K.M.; Tseng, W.-Y.; Tan, Y.-C.; Shea, K.J.; Lee, A.P. Alternating droplet generation and controlled dynamic droplet fusion in microfluidic device for CdS nanoparticle synthesis. Lab Chip 2006, 6, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Faustini, M.; Kim, J.; Jeong, G.; Kim, J.Y.; Moon, H.R.; Ahn, W.; Kim, D. Microfluidic Approach toward Continuous and Ultrafast Synthesis of Metal—Organic Framework Crystals and Hetero Structures in Confined Microdroplets. J. Am. Chem. Soc. 2013, 135, 14619–14626. [Google Scholar] [CrossRef] [PubMed]
- González-Estefan, J.H.; Gonidec, M.; Daro, N.; Marchivie, M.; Chastanet, G. Extreme downsizing in the surfactant-free synthesis of spin-crossover nanoparticles in a microfluidic flow-focusing junction. Chem. Commun. 2018, 54, 8040–8043. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Tice, J.D.; Roach, L.S.; Ismagilov, R.F. A droplet-based, composite PDMS/glass capillary microfluidic system for evaluating protein crystallization conditions by microbatch and vapor-diffusion methods with on-chip X-ray diffraction. Angew. Chem. Int. Ed. 2004, 43, 2508–2511. [Google Scholar] [CrossRef] [PubMed]
- Laval, P.; Crombez, A.; Salmon, J.-B. Microfluidic Droplet Method for Nucleation Kinetics Measurements. Langmuir 2009, 25, 1836–1841. [Google Scholar] [CrossRef] [PubMed]
- Teychené, S.; Biscans, B. Crystal nucleation in a droplet based microfluidic crystallizer. Chem. Eng. Sci. 2012, 77, 242–248. [Google Scholar] [CrossRef]
- Nightingale, A.M.; Bannock, J.H.; Krishnadasan, S.H.; O’Mahony, F.T.F.; Haque, S.A.; Sloan, J.; Drury, C.; McIntyre, R.; DeMello, J.C. Large-scale synthesis of nanocrystals in a multichannel droplet reactor. J. Mater. Chem. A 2013, 12, 4067. [Google Scholar] [CrossRef]
- Fu, Y.; Bai, L.; Zhao, S.; Zhang, X.; Jin, Y.; Cheng, Y. Simulation of reactive mixing behaviors inside micro-droplets by a lattice Boltzmann method. Chem. Eng. Sci. 2018, 181, 79–89. [Google Scholar] [CrossRef]
- Song, H.; Tice, J.D.; Ismagilov, R.F. A Microfluidic System for Controlling Reaction Networks in Time. Angew. Chem. Int. Ed. 2003, 42, 768–772. [Google Scholar] [CrossRef]
- Jiang, L.; Zeng, Y.; Zhou, H.; Qu, J.Y.; Yao, S. Visualizing millisecond chaotic mixing dynamics in microdroplets: A direct comparison of experiment and simulation. Biomicrofluidics 2012, 6, 12810–12812. [Google Scholar] [CrossRef]
- Stroock, A.D.; Dertinger, S.K.W.; Ajdari, A.; Mezić, I.; Stone, H.A.; Whitesides, G.M. Chaotic mixer for microchannels. Science 2002, 295, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Köhler, J.M.; Abahmane, L.; Wagner, J.; Albert, J.; Mayer, G. Preparation of metal nanoparticles with varied composition for catalytical applications in microreactors. Chem. Eng. Sci. 2008, 63, 5048–5055. [Google Scholar] [CrossRef]
- Wagner, J.; Tshikhudo, T.R.; Köhler, J.M. Microfluidic generation of metal nanoparticles by borohydride reduction. Chem. Eng. J. 2008, 135, 104–109. [Google Scholar] [CrossRef]
- Kirner, T.; Albert, J.; Günther, M.; Mayer, G.; Reinhäckel, K.; Köhler, J. Static micromixers for modular chip reactor arrangements in two-step reactions and photochemical activated processes. Chem. Eng. J. 2004, 101, 65–74. [Google Scholar] [CrossRef]
- Muradoglu, M.; Stone, H.A. Mixing in a drop moving through a serpentine channel: A computational study. Phys. Fluids 2005, 17, 073305. [Google Scholar] [CrossRef]
- Song, H.; Bringer, M.R.; Tice, J.D.; Gerdts, C.J.; Ismagilov, R.F. Experimental test of scaling of mixing by chaotic advection in droplets moving through microfluidic channels. Appl. Phys. Lett. 2003, 83, 4664–4666. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wang, H.; Zhang, X.; Bai, L.; Jin, Y.; Cheng, Y. Numerical simulation of liquid mixing inside soft droplets with periodic deformation by a lattice Boltzmann method. J. Taiwan Inst. Chem. Eng. 2018. [Google Scholar] [CrossRef]
- Bajer, K.; Moffatt, H.K. On a class of steady confined Stokes flows with chaotic streamlines. J. Fluid Mech. 1990, 212, 337. [Google Scholar] [CrossRef]
- Stone, H.A.; Nadim, A.; Strogatz, S.H. Chaotic streamlines inside drops immersed in steady Stokes flows. J. Fluid Mech. 1991, 232, 629. [Google Scholar] [CrossRef]
- Stone, Z.B.; Stone, H.A. Imaging and quantifying mixing in a model droplet micromixer. Phys. Fluids 2005, 17, 063103. [Google Scholar] [CrossRef]
- Nightingale, A.M.; de Mello, J.C. Segmented Flow Reactors for Nanocrystal Synthesis. Adv. Mater. 2013, 25, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Hoang, P.H.; Park, H.; Kim, D.-P. Ultrafast and Continuous Synthesis of Unaccommodating Inorganic Nanomaterials in Droplet- and Ionic Liquid-Assisted Microfluidic System. J. Am. Chem. Soc. 2011, 133, 14765–14770. [Google Scholar] [CrossRef] [PubMed]
- Paseta, L.; Seoane, B.; Julve, D.; Sebastián, V.; Téllez, C.; Coronas, J. Accelerating the Controlled Synthesis of Metal–Organic Frameworks by a Microfluidic Approach: A Nanoliter Continuous Reactor. ACS Appl. Mater. Interfaces 2013, 5, 9405–9410. [Google Scholar] [CrossRef] [PubMed]
- Frenz, L.; Blouwolff, J.; Griffiths, A.D.; Baret, J.C. Microfluidic production of droplet pairs. Langmuir 2008, 24, 12073–12076. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, A.M.; Phillips, T.W.; Bannock, J.H.; de Mello, J.C. Controlled multistep synthesis in a three-phase droplet reactor. Nat. Commun. 2014, 5, 3777. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Boedicker, J.Q.; Ismagilov, R.F. Using a Multijunction Microfluidic Device To Inject Substrate into an Array of Preformed Plugs without Cross-Contamination: Comparing Theory and Experiments. Anal. Chem. 2007, 79, 2756–2761. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Microfluidic Device | Distinctive Features | Materials Synthetized/Morphology |
|---|---|---|
| Continuous-flow | —Mixing is slow and achieved only through molecular diffusion. —This condition allows to establish controlled reaction–diffusion (RD) environments. —The diffusion of reagents is controlled in space and time avoiding chaotic advection. —A parabolic profile of fluid velocity vectors inside the microfluidic channel. —It is difficult to upscale the synthesis of materials. | Organic, inorganic, and composite materials/Continuous fibers [10,11] and continuous membranes [12,13] can be generated. |
| Segmented-flow | —Reagents can be efficiently mixed on sub-millisecond time scales via chaotic advection, and mixing can be programmed. —The no-slip boundary condition is omitted. —Consecutive reactions can be synchronized to synthetize core-shell particles and crystals. —Multiple reactions can be screened rapidly and while employing small amounts of reagents. | Organic, inorganic, and composite materials/This approach does not allow the formation of continuous fibers or continuous membranes. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonidec, M.; Puigmartí-Luis, J. Continuous- versus Segmented-Flow Microfluidic Synthesis in Materials Science. Crystals 2019, 9, 12. https://doi.org/10.3390/cryst9010012
Gonidec M, Puigmartí-Luis J. Continuous- versus Segmented-Flow Microfluidic Synthesis in Materials Science. Crystals. 2019; 9(1):12. https://doi.org/10.3390/cryst9010012
Chicago/Turabian StyleGonidec, Mathieu, and Josep Puigmartí-Luis. 2019. "Continuous- versus Segmented-Flow Microfluidic Synthesis in Materials Science" Crystals 9, no. 1: 12. https://doi.org/10.3390/cryst9010012
APA StyleGonidec, M., & Puigmartí-Luis, J. (2019). Continuous- versus Segmented-Flow Microfluidic Synthesis in Materials Science. Crystals, 9(1), 12. https://doi.org/10.3390/cryst9010012